The salmon louse is a specialist on salmonid fish. All salmonid species found along the Norwegian coast, namely Atlantic salmon (farmed and wild salmon), pink salmon, sea trout, and Arctic char, can acts as hosts for the salmon louse and thus potentially be a source of infective louse larvae. Larvae are transported with the currents and the spread can be predicted using coupled hydrodynamic-biological models, but experimental tracking of copepodids back to their host is currently not possible, and modelling is unable to distinguish between copepodids originating from farmed or wild fish in a given area. To determine origin of larvae, specific markers are needed that can distinguish between copepodids from different hosts. These markers must be reliable and easy to measure. The aim of the FHF project TraceLice (project number 90186) was to identify epigenetic markers in the genome of the salmon louse that provide information about the origin of the copepodids. DNA methylation is one of the main mechanisms in epigenetics, resulting in heritable changes in gene expression that cannot be attributed to changes in the DNA sequence and can be influenced by the environment. Epigenetic changes at known specific positions in the genome can be measured with sensitive MS-qPCR assays. To find specific significant differences in methylation patterns, adult female salmon lice from different hosts were collected along the Norwegian coast in this project. DNA methylation patterns were investigated using Reduced Representation Bisulfite Sequencing (RRBS) of these lice and analysis for consistent differences between groups of lice collected from different hosts. The same lice were also investigated for gene expression patterns using RNA sequencing. By analysing the connection between DNA methylation patterns and gene expression, the reliability of the markers identified by DNA sequencing was evaluated. Adult female lice were found on all host species, but in different numbers. Therefore, the analyses were concentrated on lice from the sea trout, Arctic char, and farmed salmon, where sufficient lice numbers were available. An important result here is that the methylation rate of the salmon louse genome turned out to be very low compared to vertebrates. Low methylation rates have also been shown for other invertebrates. Although the methylation rate of the salmon louse genome was low, several regions showed significantly different methylation rates when comparing lice from sea trout or Arctic char with lice from farmed salmon, which also showed significantly different gene expression. No single epigenetic marker could unambiguously distinguish between lice sampled from different hosts, which could possibly be solved by using several markers in combination. An epigenetic assay (MS-qPCR) was developed for one of the markers that showed significant difference. It was tested on new lice sampled from sea trout and farmed salmon in subsequent years and on their offspring. The methylation rate at this epimarker was significantly different in adult female lice from farmed salmon compared to adult female lice from sea trout. The results show that the methylation pattern of this epimarker was linked to the host species. Although the result was significant, it was not unambiguous when looking at each individual lice, which showed the need for multiple assays that together could have given an unambiguous result. The same assay was used to examine the methylation pattern in the offspring of newly collected adult female lice. The results showed that there was also a significant difference in the methylation rate of this epimarker in the copepodites. Unfortunately, the sensitivity of the method was not good enough to examine individual copepodites and could only be used for bulk measurements when many copepodites from the same egg string (siblings) were used. Further optimization of the measurement method could possibly help. The results of TraceLice show that it is potentially possible to map the origin of sea lice with respect to the host species using epigenetic markers. To be able to use use epimarkers as an analytical method would require the use of multiple markers and optimization of the measurement method for use on individual copepodites. Additional markers can be found using the data produced in the TraceLice project.
TRACELICE
— Sporing av lus ved hjelp av epigenetiske markører
Rapportserie:
Rapport fra havforskningen 2025-12
ISSN: 1893-4536
Publisert: 27.02.2025
Prosjektnr: 15993
Oppdragsgiver(e): Fiskeri- og havbruksnæringens forskningsfinansiering
Referanse: 901860
Forskningsgruppe(r):
Smittespredning og sykdom
Tema:
Lakselus
Program:
Miljøeffekter av akvakultur
Research group leader(s):
Monica F. Solberg (Smittespredning og sykdom)
Approved by:
Research Director(s):
Geir Lasse Taranger
Program leader(s):
Mari Skuggedal Myksvoll
English summary
Sammendrag
Lakselusen er en parasitt som er spesialisert på laksefisker og alle laksearter som finnes langs norskekysten, nemlig Atlantisk laks (oppdretts- og villaks), pukkellaks, sjøørret og røye kan være verter for lakselusen og kan dermed være mulige smittekilder for luselarver. Luselarver spres med strømmen og spredningen kan modelleres ved å bruke hydrodynamiske-biologiske modeller, men eksperimentell sporing av kopepoditter tilbake til deres vert er foreløpig ikke mulig, og modellering er ikke i stand til å skille mellom kopepoditter som stammer fra oppdretts- eller villfisk i et gitt område. En mulighet for å finne ut hvor parasittene stammer fra er å bruke spesifikke markører som kan skille kopepoditter fra de forskjellige artene eller skille mellom villfisk og oppdrett. Metoden må være pålitelig og relativt lett å måle. Målet med FHF prosjektet TraceLice (prosjektnummer 90186) var å identifisere epigenetiske markører i genomet til lakselusen som gir informasjon om kopepodittens opprinnelse. DNA metylering er en epigenetisk mekanisme, som kan gi varige endringer i genuttrykk som ikke kan tilskrives endringer i DNA-sekvensen og kan være påvirket av miljø. Der er forskjellige måter å måle epigenetiske endringer. En mulighet er å måle metylering ved kjente spesifikke posisjoner i genomet med sensitive metyleringsspesifikke (MS)-qPCR assayer. For å finne signifikante spesifikke forskjeller i metyleringsmønsteret i dette prosjektet ble voksne hunn-lakselus fra forskjellige verter langs Norskekysten samlet inn og studert. DNA-metyleringsmønstre ble studert ved hjelp av sekvensering av DNA fra disse lusene og analysert for konsistente forskjeller mellom grupper av lus samlet fra forskjellige verter. Av de samme lusene ble også genuttrykksmønstrene med hjelp av RNA sekvensering undersøkt. Ved å analysere samspill med DNA metyleringsmønstre og genuttrykket skulle påliteligheten til markørene identifisert ved DNA sekvenseringen evalueres. Voksne hunnlus ble funnet på alle vertsarter, men i forskjellig antall. Fokuset i analysene ble derfor på lus fra artene sjøørret, røye og oppdrettslaks. Et viktig resultat her er at metyleringsraten av lakselusgenomet viste seg å være veldig lav i forhold til vertebrater. Lav metyleringsrate har også vært vist for andre evertebrater. Det ble funnet flere genomiske regioner som viste signifikant forskjellig metyleringsrate når man sammenlignet lus fra sjøørret og lus fra røye med lus fra oppdrettslaks. Disse lusene viste også signifikant forskjellig genuttrykk. Ingen epigenetisk enkeltmarkør kunne entydig skille mellom lus fra forskjellige verter noe som muligens kunne løses ved å bruke flere markører i kombinasjon. En epigenetisk assay (MS-qPCR) ble utviklet for en av markørene som viste signifikant forskjell. Den ble testet på nye lus samplet fra sjøørret og oppdrettslaks i etterfølgende år og på deres avkom. Metyleringsraten ved denne epimarkøren var signifikant forskjellig i voksne hunnlus fra oppdrettslaks sammenlignet med lus fra sjøørret. Resultatene viser, at metyleringsmønsteret for denne epimarkøren var koblet til vertsarten. Selv om resultatet var signifikant var den ikke entydig når man så på hver enkelt lus, noe som viste behovet for flere assayer som samlet kunne gitt et entydig resultat. Samme assay ble brukt til å undersøke metyleringsmønsteret i avkom til nye innsamlede voksne hunnlus. Resultatene viste at det var en også signifikant forskjell i metyleringsraten for denne epimarkøren i kopepodittene. Sensitiviteten av metoden var dessverre ikke god nok til å undersøke enkelte kopepoditter og kunne bare brukes til bulk målinger når der bruktes mange kopepoditter fra samme eggstreng (søsken) Videre optimalisering av målemetoden ville kunne muligens hjelpe.Resultatene til TraceLice viser, at det er potensielt mulig å kartlegge opprinnelsen av lakselus med hensyn til vertsarten med hjelp av epigenetiske markører. For å kunne bruke epimarkører som analysemetode ville kreve bruk av flere markører og optimalisering av målemetoden til bruk på enkelte kopepoditter. Ytterligere markører kan finnes ved hjelp av dataene produsert i TraceLice prosjektet.
1 - Innledning
Lakselus (Lepeophtheirus salmonis) er den største utfordringen for lakseoppdrett i Norge, og avlusing forårsaker store tap for oppdrettsnæringen. Lakselus har en høy reproduksjonsrate. Fertiliserte egg deponeres i eggstrenger som bæres av hunnlusen til larvene klekkes. Antall av egg i hver sett med eggstrenger og hvor ofte disse produseres er avhengig av temperatur. På samme måte er Utviklingshastigheten av larvene er også temperaturavhengig (Hamre et al. 2019; Samsing et al. 2016). Hver sett med eggstrenger fra lakselus på laks inneholder sirka 600 egg (Thompson et al. 2023). Larvene spres med havstrømmene. Det tredje larvestadiet, kopepoditten, er det infektive stadiet. Utviklingshastigheten av larvene er også temperaturavhengig og avhengig av strøm kan larver spre seg over lange avstander (Asplin et al. 2004; Johnsen et al. 2016).Lakselusen er spesialisert på laksefisker og alle laksearter som finnes langs norskekysten (Atlantisk laks (oppdretts- og villaks), pukkellaks, sjøørret og røye) kan være verter for lakselusen og dermed være potensiell smittekilde for nye luselarver.. I motsetning til villaks som migrerer gjennom kystområdene i en begrenset tidsperiode på deres vei til storhavet, forblir sjøørret og røye i kystområdene og migrerer ikke ut i det åpne havet. De oppholder seg nær kysten (Flaten et al. 2016; Gjelland et al. 2014; Jensen et al. 2014), hovedsakelig i de øvre vannlagene hvor lakseluslarver finnes. Sjøørret kan bli i kystområdene i store deler av året, hvor de både kan bli smittet, men også sjøl bidra til smittepress. Den marine oppholdstiden for røye er 1-2 måneder (Jensen et al. 2020; Klemetsen et al. 2003), og bidraget til lakselusinfestasjon fra røye kan derfor antas å være svært begrenset da de fleste lakselus ikke vil rekke å utvikle seg til kjønnsmodne individer. Etter introduksjonen av pukkellaks i Russland, er pukkellaks nå utbredt i Norskehavet og langs norskekysten. Migrasjonsmønsteret i Norge er ikke fullt ut forstått, men det kan migrere nordover i Norskehavet nær norskekysten før det går inn i Barentshavet (Diaz Pauli et al. 2022). Pukkellakser en stillehavsart som ikke hører hjemme i Atlanterhavet. Selv om ung pukkellaks ikke er like mottakelig for lakselussmitte som atlantisk laks (Jones 2011), kan de ha betydelige infestasjoner i kystvann (Morton and Williams 2003). Tilbakevendende voksen pukkellaks er fullt mottakelig og kan bære betydelig infestasjoner (Beamish et al. 2005). Lakselusinfestasjoner av sjøørret og røye overvåkes gjennom Havforskningsinstituttet sitt nasjonale overvåkingsprogrammet for lakselus på vill laksefisk (NALO) (Nilsen et al. 2022; 2023; Serra-Llinares et al. 2014). Infisert sjøørret finnes i alle produksjonsområder (Bøhn et al. 2022; Rune et al. 2022) hvor alvorlighetsgraden av luseangrep er korrelert til lokal lakseoppdrettsintensitet (Bøhn et al. 2022). Men mens oppdrettslaks avluses i henhold til regulatoriske grenser, kan antallet lus på sjøørret være hundrevis i de sterkt infiserte områdene (Gjelland et al. 2014) med høy mengde voksne lus (Vollset et al. 2018). Også på pukkellaksen har det blitt registrert lakselus (Lorentzen 2023), men antall voksne hunnlus var relativt lav. Spredningen av luselarver kan forutses ved å bruke koblede hydrodynamiske-biologiske modeller (Asplin et al. 2020). Imidlertid er eksperimentell sporing av det infektive stadiet kopepoditter tilbake til deres opprinnelse foreløpig ikke mulig, og modellering er ikke i stand til å skille mellom kopepoditter som stammer fra oppdretts- eller villfisk i et gitt område. Det har vært forsøk på å identifisere markører for kopepoditter av ulik opprinnelse, for eksempel i FHF prosjekt nr. 900790, og den nettopp rapporterte oppfølgende prosjekt (FHF nr. 901859) hvor forskjellige markører, avhengig av den kjemiske sammensetningen til verten til lusen, ble brukt. Disse må imidlertid fortsatt verifiseres i TraceLice prosjektet skulle det undersøkes mulige epigenetiske forskjeller i lakselus avhengig av opprinnelsesvert og om disse forskjellene kan brukes som opprinnelsesmarkører. Epigenetikk er et raskt voksende forskningsfelt, men foreløpig er det begrenset med studier av virvelløse dyr (Klughammer et al. 2023). DNA-metylering er en epigenetisk modifikasjon som innebærer tilsetning av en metylgruppe til en cytosinbase i DNA (5-metyl-cytosin (5-mC)). Endringer i DNA-metyleringsmønstre kan ha funksjonelle konsekvenser for genuttrykk, cellulære prosesser og utvikling av fenotype (Piferrer and Wang 2023). DNA-metyleringsmønsteret kan endres på grunn av miljøpåvirkninger (Feil and Fraga 2012). Hos virveldyr blir spesielle regioner i promotorregionen av genene metylert, og hindrer bindingen av transkripsjonsfaktorer og derved hemmer at genet kan leses av. Hos virvelløse dyr som lakselus er det funnet et annet mønster hvor exoner og introner av visse gener er metylert (Feng et al. 2010; Keller et al. 2015). Her kan metylering av gener korreleres med genuttrykk, omvendt til det som er kjent fra virveldyr. Uavhengig av den dempende eller stimulerende funksjonen til epigenetiske markører, kan de oppstå i én generasjon og videresendes til avkommet (Skjærven et al. 2023). Derfor kan markører som oppstår i et vertsavhengig mønster og nedarves i avkommet være en effektiv indikator på opprinnelse. Fenotypisk plastisitet, evnen at en genotype kan produsere forskjellige fenotyper som en funksjon av miljøforhold, kan være et resultat av epigenetiske endringer. Epigenetiske markører kan gå fra foreldre til avkom gjennom epigenetisk arv. Dette er funnet i et bredt spekter av arter, inkludert det lille planktoniske krepsdyret Daphnia, (VandegehuchteDe Coninck et al. 2010; VandegehuchteLemière et al. 2010). Også i kopepoden Paracyclopina nana er der vist multigenerasjons epigenetisk plastisitet mot havforsuring (Lee et al. 2022). I lakseluskopepoditter ble der funnet endringer i den totale 5-methylcytosine nivåer i henhold til ferskvannsbehandling (FHF prosjekt OptiDeLouse: Prosjektnummer 901687). I TraceLice prosjektet skulle denne mekanisme som dyr bruker til å justere genetikken sin uten å endre DNA sekvensen utnyttes til å påvise arvelige forskjeller mellom lus fra forskjellige verter. DNA metyleringsmønstre kan måles i genomet ved hjelp av «Reduced Representation Bisulfite Sequencing» (RRBS). For å utføre RRBS kuttes genomisk DNA med restriksjonsenzymer som deretter bisulfitt behandles. Bisulfittbehandling konverterer umetylerte cytosin til uracil, mens de metylerte cytosiner bevares. Deretter sekvenseres DNAet for å identifisere metylerte og ikke metylerte cytosinrester ved spesifikke områder i genomet. For å bestemme metyleringstilstanden ved kjente spesifikke steder på DNA kan man bruke metyleringsspesifikke qPCR assayer (MS-qPCR), en qPCR-teknologi for kvantifisering av metylering med hjelp av spesifikke primere.
Målet med TraceLice var å identifisere epigenetiske markører i genomet til lakselus som gir pålitelig informasjon om kopepodittens opprinnelse. Dette ble adressert gjennom følgende delmål:
Delmål 1: Identifisere vertsspesifikke (hvilken fiskeart) markører i lakselus. Dette gjøres ved å undersøke DNA-metyleringsmønstre ved hjelp av sekvensering av voksne hunnlus fra ulike verter og analysere disse for konsistente forskjeller.
Delmål 2: Evaluere påliteligheten til markørene identifisert i delmål 1 ved å analysere deres samspill med genuttrykket av relevante gener. Dette gjøres ved å RNA-sekvensere lusene og så innlemme de resulterende genuttrykksmønstrene med DNA-metyleringsdataene for å evaluere epimetyleringsmarkørene identifisert i delmål 1.
Delmål 3: Utvikle og teste en analysemetode som identifiserer hvilken fiskeart kopepodittene ble produsert fra. For å gjøre dette skal vi utvikle metyl-spesifikk qPCR-assayer (MS-qPCR) rettet mot epimetyleringsmarkørene identifisert i delmål 1.
2 - Prosjektets omfang
Totalbudsjettet var på 2.46 mill. NOK, og fullfinansiert av FHF. Prosjektperioden var 14 måneder.
3 - Prosjektorganisering
Prosjektet ble utført med Universitet i Bergen som prosjekteier, og Havforskningsinstituttet som samarbeidspartner. Christiane Eichner var prosjektleder. FHF representant var Kjell Maroni.
Havforskningsinstituttet har hatt ansvar for innsamling av prøvemateriale, for DNA metyleringsanalysen med hjelp av Reduced Representation Bisulfite Sequencing» (RRBS) og for utviklingen av en metyleringsspesifikk (MS)-qPCR assay samt analyse av prøver med denne.
Universitet i Bergen har hat ansvar for RNA sekvenseringen, fotografering og oppdeling av prøvematerialet og klekking av lakseluslarver.
Aktiviteten i prosjektet var inndelt i fem arbeidspakker (AP) som vist nedenfor, som hver danner grunnlag for den neste basert på oppnåelse av spesifikke mål. Unntak var AP5 (prosjektstyring) som var ansvarlig for koordinasjon, rapportering og kommunikasjon av prosjektet.
AP1: Prøvetaking av lus
Ansvarlig: Havforskningsinstituttet, Rasmus Skern Mauritzen og Sussie Dalvin
AP2: Bestemmelse av differensielt metylerte regioner
Ansvarlig: Havforskningsinstituttet, Takaya Saito
AP3: Bestemmelse av differensielt uttrykte gener ved hjelp av RNA sekvensering
Ansvarlig: Universitet i Bergen, Christiane Eichner
AP4: Implementering av en MS-qPCR-assay for undersøkelse av utvalgte metyleringssteder av interesse
Ansvarlig: Havforskningsinstituttet, Kaja Skjærven og Kai Ove Skaftnesmo
AP5: prosjektstyring
Ansvarlig: Universitet i Bergen, Christiane Eichner
Det overordnet mål for arbeidspakkene var å utvikle en analysemetode som kan brukes til å skille mellom lus av forskjellig vertsopprinnelse. Et grafisk sammendrag av prosjektorganiseringen er vist i Figur 1.

4 - Problemstilling og formål
Lakselusen er den største enkeltutfordringen for norsk oppdrettsnæring; og forårsaker store direkte og indirekte kostnader knyttet til tiltak for bekjempelse av lakselus og forringelse av fiskevelferd. Lusekontroll er derfor av stor betydning. Et uunnværlig element i bekjempelsen av lakselus er å forebygge nye angrep, noe som avhenger av at man kjenner kilden til ny smitte. Derfor hadde TraceLice som mål å utvikle et verktøy for å knytte smittsomme kopepoditter til deres opprinnelse i form av oppdrettslaks eller ville laksefisk.
4.1 - Resultatmål
I løpet av prosjektet ble der foretatt grunnleggende analyser av epigenetiske variasjoner i lakselusen for å se om disse sammenfalt med opphavet til lakselusen. Dette førte til identifisering av en rekke markørkandidater. Disse markører ble også bekreftet ved å studere om forskjeller med lakselus fra ulike fiskearter var gjenspeilet i ulik genekspresjon. Basert på disse vurderinger ble der utviklet to forskjellige assayer designet til å kunne spore opphavet til lakselusen. Dessverre ga ingen av de utvalgte assay alene et svar, men det vurderes at man med noe mer optimalisering av prøvebehandling (for kopepoditter) og mindre metodiske justeringer i fremtiden kan finne opphav til lakselus på oppdrettsfisk.
I tillegg har prosjektet generert data om epigenetikk generelt i en dyregruppe som er lite undersøkt og kan danne grunnlag for en rekke nye studier av lakselus og deres evne til tilpasning som respons til eksempel endret miljø eller behandlinger.
5 - Prosjektgjennomføring og oppnådde resultater
Prosjektet ble gjennomført som planlagt med første samplinger i sommeren innen prosjektoppstart. På grund av noen forsinkelser i siste arbeidspakke ble prosjektet gjennomført med 2 måneders forlengelse.
5.1 - Arbeidspakke1: Prøvetaking av lus
Mål: skaffe både voksne hunnlus og kopepoditter av forskjellig opprinnelse
Innsamling av voksne hunnlus og eggstrenger med forskjellig vertsopprinnelse ble utført i forbindelse med vitenskapelig undersøkelsestrål og Havforskningsinstituttets luseovervåkingsprogram langs norskekysten (NALO) (Nilsen et al. 2020) . Lus fra pukkellaks ble samplet fra fisk fanget av lokale fiskere ved Bugøynes. Lusene ble innsamlet i to omganger: 2023 for å finne differensielt metylerte steder i genomet til voksne hunnlus fra forskjellige vertsfiskearter gjennom genom- og transkriptom- analyse (AP2 og AP3) og 2024 for å verifisere disse steder med uavhengige lus og deres avkom (AP4). I 2023 ble voksne hunnlus fiksert dirkete i RNA later mens i 2024 ble levende lus med eggstreng innsamlet og transportert til laboratoriet hvor lus ble fiksert på RNA later og eggene ble klekket i laboratoriet for analyse av kopepoditter (AP4).
I 2023 ble i alt 124 lus samplet og fiksert for dette prosjektet som videre ble brukt i AP2 og AP3. Alle kom fra ruse/garn fanget fisk. Alle lusene ble tatt fra forskjellige fisk for å fokusere på vertspesifikke forskjeller og e ikke eventuelle forskjeller mellom individuelle fiskt. Av disse var: 82 lus samplet fra sjøørret, 11 lus samplet fra røye, 3 lus samplet fra villaks, 2 lus samplet fra pukkellaks. I tillegg ble 25 voksne hunn-lus samplet fra oppdrettslaks ved Austevoll Havbruksstasjon fra to forskjellige merder ved forskjellige dager. Andel lus samplet fra de forskjellige ville vertene var som i tidligere år (Nilsen et al. 2022) med flest sjøørret og få villaks.. På grunn av den korte tiden som villaks bruker til å vandre gjennom fjordene til havet, finnes der sjeldent voksne hunnlus på utvandrende villaks, men bare yngre stadier, og den tilbake vandrende villaks samples ikke grunnet vern. Etter innsamlingen i 2023 ble alle RNA later fikserte adulte hunnlus inspisert under lupen, fotografert, målt og delt opp med hjelp av skalpell for videre analyse. Forkroppen ble delt på langs i to like halvdeler (Figur 2a) for henholdsvis DNA metyleringsanalyse (AP2) og genuttryksanalyse (AP3). Lusene ble målt og forholdet mellom lengden på forkroppen og totallengden ble bestemt for å undersøke modningsgraden. Lengden av lusene varierte (Figur 2b).

Ikke alle voksne lus som ble innsamlet var fullt modent, som vises ved et ikke fullstendig utvokst genitalsegment (Eichner et al. 2008). Siden modningsgraden har innvirkning på genuttrykket (Eichner et al. 2008) ble bare full modne lus brukt til RNA sekvenseringen. I de tilfellene hvor ikke et tilstrekkelig antall full modne lus var tilgjengelig, ble også voksne ikke full modne lus brukt for å analysere metyleringsmønsteret, men ikke for genuttrykket. Siden antall lus fra villaks som forventet var lavt (2 full modne voksne lus og en ikke full moden voksen lus) ble denne gruppen supplert med lus samplet fra villaks til annet formål i et tidligere år. Disse var fiksert på etanol og kunne derfor bare brukes til DNA-, men ikke RNA-sekvenseringen siden RNA raskt degraderes ved fiksering på etanol. I tillegg til lus innsamlet i felt ble ti lus og deres avkom fra et laboratoriet eksperiment inkludert. Her var halvparten av disse lus, hadde blitt utsatt for ferskvannsbehandling. Resultatene ble sammenlignet med samplene fra felt til å undersøke påvirkning av ferskvann. En oversikt over alle analyserte lus og dens opprinnelse (produksjonsområde) er vist i Tabell 1.
| Antall lus til | |||
| Vertsart | PO | DNA metyleringsanalyse | Genuttrykk (RNA) |
| Oppdrettslaks | 3 | 8 | 7 |
| Oppdrettslaks | 3 | 8 | 8 |
| Sjøørret | 2 | 5 | 5 |
| Sjøørret | 2 | 3 | 3 |
| Sjøørret | 5 | 6 | 3 |
| Sjøørret | 9 | 8 | 8 |
| Sjøørret | 13 | 6 | 5 |
| Pukkellaks | 13 | 2 | 2 |
| Røye | 10 | 7 | 6 |
| Røye | 9 | 1 | - |
| Røye | 11 | 1 | - |
| Villaks | 11 | 1 | 1 |
| Villaks | 4 | 1 | 1 |
| Villaks | 10 | 1 | - |
| Villaks | 4 | 8* | - |
| Villaks | 5 | 8* | - |
| Oppdrettslaks laboratorium SW | - | 5 | 5 |
| Oppdrettslaks laboratorium FW | - | 5 | 5 |
| SUM | 84 | 59 | |
Levende lus med eggstreng ble samplet i 2024. Disse ble brukt i AP4 til å verifisere resultatene som ble hentet fra dataene fra 2023 lusene med hjelp av en metyleringsspesifikk-qPCR assay. I tillegg til å undersøke disse uavhengige prøver ble også avkommet deres undersøkt. Lusene ble samplet fra sjøørret (Etne) og fra oppdrettslaks (Matre og Fensfjorden). Lusene ble lagt levende hver for seg transportert til UiB, i 50ml Falcon rør, hvor lusene ble fiksert og eggene ble parvis lagt til klekking i nummererte klekkebrønner med gjennomstrømmingsvann. I alt ble 129 lus med eggstrenger samplet, 92 fra sjøørret og 37 fra oppdrettslaks. Klekking av egg og utvikling til kopepoditter ble observert for å ta prøvene av larver med definert alder. Dyrene ble prøvetatt 3,5 dager etter skallskifte til kopepoditt. Alle kopepodittene ble undersøkt under lupen før fiksering og en del ble fotografert. Ikke alle egg klekket, muligens grunnet dårlig vannkvalitet i transportrørene. Disse ble fylt med vann fra fjorden og saliniteten kan ha vært lav. Bare prøver med tilsynelatende friske kopepoditter ble analysert. 20 lus og deres avkom fra sjøørret og 23 lus og deres avkom fra oppdrettslaks.
5.2 - Arbeidspakke 2: Undersøkelse av DNA metylering
Mål: kartlegge differensielt metylerte steder mellom vill og oppdrettet laksefisk som startpunktet for implementeringen av en MS-qPCR-analyse
Ved hjelp av «Reduced Representation Bisulfite Sequencing» (RRBS) av voksne hunnlus, fra forskjellige verter og regioner, ble DNA metylerings mønstre kartlagt. Basert på sekvensdataene ble en differensiell metyleringsanalyse utført for å identifisere områder med ulik metylering avhengig av vertsfisk.
Til dette formål ble DNA renset fra halvparten av forkroppen og brukt til RRBS sekvensering (Illumina HiSeq) ved «Genomics Core Facility» (UiB). Sekvenseringen produserte e i gjennomsnitt 48 millioner sekvenser per prøve. RRBS sekvensene ble alignet mot lakselus (Lepeophtheirus salmonis salmonis) genomet Assembly: LSalAtl2s, 54 (Ensembl) (Skern-Mauritzen et al. 2021). I gjennomsnitt alignet 74% av sekvensene unikt. CpG-steder, ble ekstrahert og bisulfittkonverterte sekvenser kartlagt og cytosinmetyleringstilstander bestemt. Forskjeller i metyleringsmønstre mellom lus fra forskjellig opprinnelse ble undersøkt. Til dette formål ble ikke bare lus fra forskjellige verter sammenlignet mot hverandre, men også andre grupperinger ble undersøkt for å studere påvirkningen av geografisk område (nord versus sør, forskjellige PO) eller mulig ferskvannspåvirkning og til å finne spesifikke mønstre i prøvematerialet. Både antall hypo- og hyper-metylerte posisjoner ved sammenligning av forskjellige grupper og deres posisjonering i genomet ble undersøkt. I tillegg ble det undersøkt i hvilke regioner i genomet (for eksempel exon, intron, promotor) metyleringen er funnet og om der var forskjeller i mønsteret.
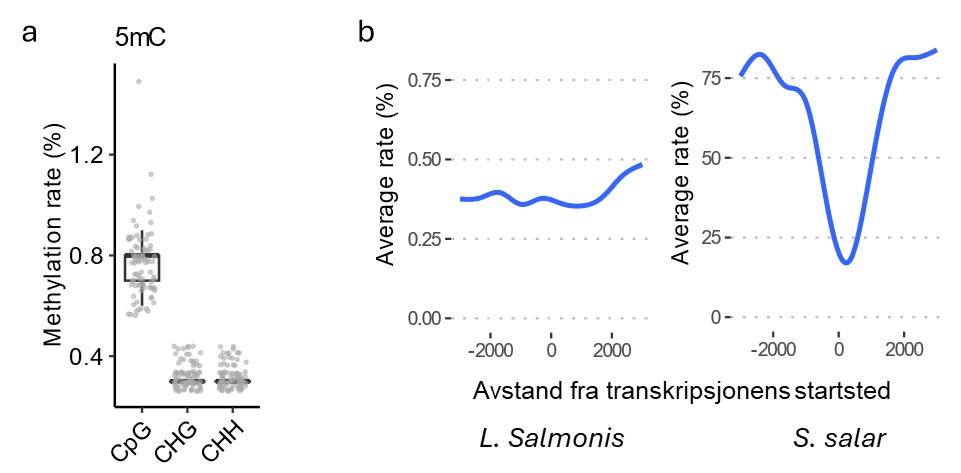
Generelt viste lusens sitt genom å være veldig lite metylert (Figur 3a). Sirka 0,8% av CpG steder var metylert, men dette er ikke uvanlig for genomer av virvelløse dyr hvorimot metyleringsraten i virveldyr kan være 70-80% (Figur 3b). Metyleringen i lakselus var jevnt fordelt over genomet og viser ikke noe spesifikt mønster i forhold til transkripsjonsstarten som man ser for eksempel i laksens genom (Figur 3b).
Lus fra villaks, som var fiksert på etanol (opprinnelig samplet til andre formål) skilte seg ut fra alle andre prøvene, også fra lus samplet fra villaks for dette prosjektet og fiksert på RNA later (bare 3 prøver). Siden vi ikke kunne være sikkert på om dette skyldes de forskjellige fikseringsmetoder eller om andre omstendigheter var årsaken, ble samplene ikke tatt med i videre analyse. Videre konsentrerte vi alle følgende analyser på lus fra oppdrettslaks, sjøørret og røye til å få statistisk sterke resultater, siden antall prøver fra villaks og pukkellaks var for lavt (2 prøver fra pukkellaks og 3 prøver fra villaks, hvorav en var umoden og kunne ikke brukes til RNA sekvensering). Antall og opprinnelse av prøvene brukt til videre analyse er vist i Figur 4.

Metyleringsmønstre mellom prøver samplet fra oppdrettslaks ble sammenlignet med metyleringsmønstre av prøvene samplet fra sjøørret og røye. Resultatene fra denne differensielle metyleringsanalysen er vist i Figur 5. Vi finner både differensielt metylerte cytosiner (DMCs) som er hyper- og hypo-metylert i oppdrettslaksen sammenlignet med de ville artene sjøørret eller røye. Videre analysene i samspill med resultater fra AP3 er beskrevet lenger ned sammen med resultatene fra AP3.

Dataene er tilgjengelig i Sequence Read Archive (SRA) med følgende referansenummer: PRJNA1071881.
5.2.1 - Påvirkning av ferskvann på metyleringsmønstre
Lakselus på fisk i fjorden kan i perioder utsettes for fersk og brakkvann. For å undersøke denne påvirkningen ble voksne hunnlus fra et laboratorieeksperiment undersøkt. Atlantisk laks ble smittet med lakselus og en gruppe gikk ved reduserte saliniteter i to generasjoner (27 og 24 ppt i hhv. 1. og 2. generasjon) og ble ytterligere behandlet med ferskvann 3 ganger pr. generasjon. En kontrollgruppe ble ikke utsatt for dette behandling. Metyleringsmønstre mellom avkommet fra lus fra de to gruppene ble sammenlignet; de analyserte individer har dermed ikke selv blitt utsatt for ferskvann og redusert salinitet. Forskjeller i den samlete genuttrykk vises ved prinsipalkomponentanalyse (PCA) plot i Figur 6a. Lusene skiller seg i sin overordnete metyleringsmønster. Alle differensielt metylerte cytosiner (DMCs) er hypo-metylert i lus som var utsatt for ferskvannsbehandling (Figur 6b, c).

5.3 - Arbeidspakke 3: RNA sekvensering
Mål: evaluere påliteligheten til markørene identifisert i AP2 ved å analysere deres assosiasjon med uttrykk av relevante gener.
I denne AP ble den totale genuttrykk av de samme lusene som ble undersøkt i AP2 undersøkt ved hjelp av RNA sekvensering. Formålet med dette var å finne gener som er påvirket i sitt uttrykk av differensiell metylering. Dette ble gjort ved å sammenligne uttrykk av gener (RNA sekvensering) med DNA-metyleringsresulter oppnådd i AP2.
RNA av den andre halvparten av forkroppen til 59 voksne hunnlus (Tabell 1) ble renset og mRNA ble sekvensert (Illumina HiSeq). ved «Genomics Core Facility» (UiB) og resulterte i gjennomsnittlig 32 millioner sekvenser fra hver prøve. Sekvensene ble alignet mot lakselus genomet (LSalAtl2s). Av disse var gjennomsnittlig 88% «lusegener», altså sekvensen som kan gjenfinnes i genomet til lakselus. I tillegg ble også kopepodittene, som var avkom til lusene fra laboratorieeksperiment, sekvensert for å se om endringer i genuttrykk ble overført til avkom. Forskjell i den samlete genuttrykk vises ved prinsipalkomponentanalyse (PCA) plot i Figur 7. Den samlete genuttrykk av lusen adskiller ikke lus samlet fra oppdrettslaks, villaks og sjøørret, men den samlete genuttrykk fra lus fra røye skiller seg ut. Også lus fra oppdrettslaks holdt i laboratoriet adskiller seg fra de andre prøvene.

Videre ble de relevante prøvene fra oppdrettslaks, sjøørret og røye analysert. Figur 8a viser en sammenligning av den overordnete genuttrykk. Genuttrykket til lus samplet fra røye skiller seg ut. 4423 gener var signifikant forskjellig regulert i sjøørret i forhold til oppdrettslaks (2435 opp, 1988 ned-regulert) og 5420 gener var signifikant forskjellig regulert i røye i forhold til oppdrettslaks (2696 opp-, 2724 ned-regulert) (Figur 8b). Selv om der er mange gener som er sterkt signifikant regulert mellom gruppene er de fleste av dem bare svakt regulert (mindre enn 1,5 ganger øking eller reduksjon) (Figur 8b).

Genuttrykket i lus samplet fra de forskjellige vertene til noen av de signifikant forskjellig uttrykte genene er vist i Figur 9.

5.3.1 - Påvirkning av ferskvann på genuttrykk
Lusene fra ferskvannsforsøk ble undersøkt for genuttrykket med hjelp av RNA sekvensering. Lusene fra de to gruppene skiltes tydelig i sitt overordnete genuttrykk (Figur 10a). Eggstrengene til disse voksne hunnlusene ble klekket og genuttrykket ble også målt i dens avkom, i kopepodittene. Også kopepodittene fra de to gruppene kunne skilles tydelig i sitt overordnete genuttrykk (Figur 10b). Analysen viste også at forskjellene i genuttrykk varierte mellom voksne lus og deres avkom, slik at forskjellene mellom ferskvannsbehandlet og kontroll lus var tydelig, men det var bare 27 (15 +12) transkripter som var regulert i både voksne og avkom (Figur 10c).

5.3.2 - Påvirkning av ferskvann på metylering og genuttrykk
Vi finner mange gener (233), som er både signifikant forskjellig uttrykt i voksne hunnlus og viser et signifikant forskjellig metyleringsmønster ved posisjonen i eller ved samme gen (Figur 11). Når man i tillegg sammenligner genuttrykket i avkommet deres, finner vi 11 gener, som er regulert i alle tre grupper. Hvordan DNA er metylert i kopepodittene, vet vi ikke (samplene ble bare brukt til RNA sekvenseringen) og vi kan derfor ikke si at der er en sammenheng.

Dataene er tilgjengelig i Sequence Read Archive (SRA) med følgende referansenummer: PRJNA1090296
5.4 - Valg av spesifikke metyleringssteder basert på resultater fra AP2 og AP3
Resultatene fra metyleringsanalysen (AP2) og fra genuttrykksanalysen (AP3) ble kombinert og differensiell uttrykte gener, som inneholder flere differensielle metyleringssteder ble valgt. I Tabell 2 er de 8 beste kandidatgener vist. Disse er enten bare regulert i røye, sammenlignet med oppdrettslaks (2 gener), i sjøørret, sammenlignet med oppdrettslaks (1 gen), eller i begge (5 gener).
| Røye | Sjøørret | ||||||||||
| GenID | regulert | Exoner | Introner | Prom | Flanker | regulert | Exoner | Introner | Prom | Flanker | Beskrivelse |
| 931 | Opp | 0 | 0 | 11 | 3 | Opp | 0 | 0 | 8 | 5 | Peptidyl-prolyl cis-trans isomerase |
| 12703 | Ned | 0 | 9 | 0 | 0 | Ned | 0 | 11 | 0 | 0 | UDP-N-acetylglucosamine/UDPglucose/GDP-mannose transporterlike isoform X1 |
| 8333 | Opp | 0 | 5 | 0 | 0 | Opp | 0 | 5 | 0 | 0 | Flightin-like protein |
| 6059 | - | 0 | 0 | 0 | 0 | Ned | 0 | 10 | 0 | 0 | Protein angel homolog 2 |
| 5407 | Ned | 0 | 0 | 0 | 4 | Ned | 0 | 0 | 0 | 5 | Ca(2+)/calmodulin-responsive adenylate cyclase isoform X2 |
| 7605 | Ned | 5 | 3 | 0 | 0 | - | 0 | 0 | 0 | 0 | Neurofilament heavy polypeptide |
| 7648 | Opp | 0 | 8 | 0 | 0 | - | 0 | 0 | 0 | 0 | bestrophin-4 isoform X1 |
| 7651 | Opp | 0 | 4 | 0 | 0 | Opp | 0 | 4 | 0 | 0 | Troponin C, isoform 1 |
I tillegg ble videre mulige DNA metyleringsmarkører identifisert (Tabell 3 og Figur 12) ved bruk ave de statistiske metodene glmnet (Lasso and Elastic-Net Regularized Generalized Linear Models) (Friedman et al. 2010) og t-scores eller ved hjelp av methylKit (Akalin et al. 2012). Selv om metyleringsraten ved de viste posisjoner er signifikant forskjellig mellom lus fra de forskjellige undersøkte vertene ser vi en del overlapp og ingen av markørene viser et entydig mønster.

| Vert | Metode | Scaffold | Start | Ende | Strand | DMC | GenID | Region | Beskrivelse |
| AC | glmnet | LSalAtl2s254 | 384933 | 384933 | + | Hyper | 4826 | Exon | trypsin-1-like |
| 4825 | P250 | PRSS1_2_3 | |||||||
| 4827 | P5K | trypsin-1-like | |||||||
| LSalAtl2s1402 | 66035 | 66035 | - | Hypo | - | - | - | ||
| LSalAtl2s163 | 480304 | 480304 | - | Hyper | 2840 | Intron | band 4.1-like protein 4 isoform X1 | ||
| t score | LSalAtl2s16 | 435676 | 435676 | + | Hypo | 2957 | P5K | somatomedin-B and thrombospondin type-1 domain-containing protein | |
| LSalAtl2s290 | 636836 | 636836 | + | Hypo | 5408 | Flank | Zinc finger C2H2-type,Zinc finger, RING/FYVE/PHD-type | ||
| LSalAtl2s1113 | 114669 | 114669 | + | Hypo | - | - | - | ||
| ST | glmnet | LSalAtl2s16 | 435676 | 435676 | + | Hypo | 2957 | P5K | somatomedin-B and thrombospondin type-1 domain-containing protein |
| LSalAtl2s1402 | 113433 | 113433 | - | Hypo | 2189 | Flank | protein LSM12 homolog | ||
| LSalAtl2s273 | 541158 | 541158 | + | Hyper | 5186 | Flank | homeobox protein MSH-Dlike isoform X3 | ||
| t score | LSalAtl2s542 | 403586 | 403586 | + | Hypo | - | - | - | |
| LSalAtl2s967 | 1312 | 1312 | + | Hypo | 12742 | Flank | DNA-binding protein HEXBP | ||
| LSalAtl2s23103 | 316 | 316 | - | Hypo | - | - | - | ||
| AC&ST | methylKit | LSalAtl2s290 | 636791 | 636791 | + | Hypo | 5408 | Flank | Zinc finger C2H2-type, Zinc finger, RING/FYVE/PHDtype |
| 636834 | 636834 | + | Hypo | - | - | - | |||
| 636836 | 636836 | + | Hypo | - | - | - | |||
| 636854 | 636854 | + | Hypo | - | - | - | |||
| LSalAtl2s696 | 230081 | 230081 | + | Hypo | 10481 | Flank | Pre-mRNA-splicing factor RBM22 | ||
| 10482 | Flank | E3 ubiquitin-protein ligase KCMF1-like | |||||||
| 230090 | 230090 | + | Hypo | 10481 | Flank | Pre-mRNA-splicing factor RBM22 | |||
| 10482 | Flank | E3 ubiquitin-protein ligase KCMF1-like | |||||||
| DEG&DMC | LSalAtl2s1162 | 152386 | 152386 | - | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase | |
| 152395 | 152395 | - | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase | |||
| 152496 | 152496 | + | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase | |||
| 152497 | 152497 | - | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase | |||
| 152867 | 152867 | - | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase | |||
| 152880 | 152880 | + | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase | |||
| 152881 | 152881 | - | Hypo | 931 | P5K | Peptidyl-prolyl cis-trans isomerase |
5.5 - Arbeidspakke 4: Design av en diagnostisk assay basert på artsspesifikke metyleringssteder
I denne arbeidspakke ble en metyleringsspesifikk qPCR (MS-qPCR) assay etablert for å undersøke og validere DNA-metyleringsendringene i spesifikke gener eller genregioner oppdaget i voksne hunnlus i AP2 og AP3 til bruk i enkelte kopepoditter. MS-qPCR kan gi en kvantitativ vurdering av DNA-metyleringsendringer på spesifikke steder i genomet. Dens anvendelse for å påvise metyleringstilstanden på disse spesifikke stedene ble testet i nye voksne hunnlus innsamlet sommeren 2024 (AP1) og i kopepoditter klekket fra eggstrenger av disse lusene.
Mål: Lage en MS-qPCR assay for å undersøke metyleringsstatus i spesifikke gener som ble funnet i AP2 og AP3 til å kunne diskriminere mellom lus fra ulike fiskearter.
En mulig metode for å validere endringer i DNA-metylering i spesifikke gener eller regioner er å designe primere som skiller mellom metylerte og umetylerte sekvenser, og deretter bruke RT-qPCR til å måle genuttrykket. Denne metoden kalles metyleringsspesifikk qPCR (MS-qPCR) og er en svært sensitiv metode som kan utføres med svært lite utgangsmateriale.
MS-qPCR kan gi en kvantitativ vurdering av DNA-metyleringsendringer på spesifikke CpG-steder. Det første trinnet i metoden er å bruke bisulfittbehandling for å omdanne umetylerte cytosiner til uracil, mens metylerte cytosiner forblir uendret. Deretter designes primere som spesifikt amplifiserer enten metylerte eller ikke metylerte sekvenser, og qPCR utføres for å måle mengden amplifisert produkt for hver type sekvens. Ved å sammenligne nivåene av metylerte og ikke metylerte produkter kan graden av DNA-metylering kvantifiseres. Men det er ikke bare en utfordring å finne spesifikke steder, som kan brukes til å skille opphav av lusene, men DNA regionen rundt disse stedene må i tillegg være egnet til å konstruere spesifikke primere. To forskjellige markører ble valgt (Figur 13). Begge posisjoner viser signifikante forskjeller i metyleringsraten mellom lus fra sjøørret sammenlignet med lus fra oppdrettslaks, men er ikke entydig.

Spesifikke primere til både markører kunne lages, men det viste seg, at assayen, som skulle måle metylering i marker 2 ikke virket. Men assayen til marker 1 kunne skille mellom forskjellig metylert cytosin ved den valgte posisjon, som standardkurven viser (Figur 14a). Voksne hunnlus samplet i 2024 (AP1) fra sjøørret og oppdrettslaks ble undersøkt for metyleringsmønsteret ved posisjon til markør 1 med hjelp av den utviklete assayen. Nye lus ble brukt til å validere funnene fra DNA sekvenseringsanalysen gjennomført med lus samplet i 2023. Med å undersøke nye lus forsikrer man seg at markøren virkelig skiller mellom lus samplet fra sjøørret sammenlignet med lus samplet fra oppdrettslaks og at ingen bias i prøvene fra 2023 er ansvarlig for resultatet. Resultatene er vist i Figur 14b. Lusene var signifikant forskjellig i metyleringsraten av cytosin ved dette posisjon og kunne dermed verifisere funnene fra 2023. Men som forventet, kunne metyleringsraten ikke skille lusene fra de forskjellige vertene entydig fra hverandre. En mulig løsning til dette problemet kunne være å lage flere assayer, som til sammen viser et entydig resultat. Analysene vist til nå ble gjord ved voksne hunnlus av kjent opphav. I siste skritt skulle det undersøkes, om voksne hunnlusene overfører metyleringsmønster ved den undersøkte posisjonen til avkommene sine og om metoden er egnet til å skille kopepoditter fra mødrene fra forskjellige verter. For dette formål ble metyleringsraten ved markør 1 målt i kopepodittene som var avkom av lusene samplet i 2024 og målt med MS-qPCR assayen. Metyleringsraten ble målt på enkelte kopepoditter og på flere kopepoditter fra samme mor (batch med 100 til 200 kopepoditter). Resultatene for batch målingene av kopepoditter er vist i Figur 14c.

Der er en signifikant forskjell i metyleringsraten ved posisjonen av markør 1 i kopepodittene som er avkom av mødrene fra sjøørret sammenlignet med mødrene fra oppdrettslaks. Dessverre er resultatene som forventet ikke entydig her heller. Måling av metylering i enkelte kopepoditter var med den anvendte metoden dessverre under deteksjonsgrensen. Resultatene viser, at det er prinsipielt mulig å bruke metyleringsraten av cytosin ved spesifikke posisjoner til å skille lus fra forskjellig opphav. Men til å kunne anvende metoden kreves flere optimaliseringer. Der trenges flere assayer, som kunne utvikles basert på resultatene innhentet i AP2 og AP3, som i en helhet ville gi et entydig resultat. Dessuten viste assayen seg å ikke være sensitiv nok til å måle metylering i enkelte kopepoditter. Fiksering av kopepodittene på RNA later er muligens ikke optimalt, men ble brukt til å være konsekvent i anvendelse av fikseringsmetoden. Å bruke RNA later var nødvendig for lusene samplet i 2023 til bruk for DNA og RNA sekvensering, men DNA utbytte i etanolfikserte prøver er vanligvis bedre. Optimalisering av selve assayen kunne også hjelpe.
6 - Diskusjon og konklusjon
I dette prosjektet kunne det vises ulike metyleringsmønstre i DNAet fra lus plukket fra oppdrettslaks sammenlignet med lus fra sjøørret eller røye og dermed at det er prinsipielt mulig å bruke metyleringsraten av cytosin til å skille lus fra forskjellig opphav. Disse forskjellene var statistisk signifikante, men prosjektet fant ingen enkelposisjon som alene kan identifisere opphav. Selv om største delen av lusene fra en gruppe viste høyere eller lavere metyleringsraten ved den spesifikke CpG posisjonen i forhold til lus fra den andre gruppen, var der lus som ikke passet inn i mønsteret. Dette skyldes sannsynligvis blant annet, at metyleringsraten i lakselus er generelt veldig lav og dermed forskjeller i metyleringsraten små. Metyleringsraten i genomet til lakselusen var ikke kjent fra før og er også et resultat oppnådd it dette prosjektet. Sannsynligvis vil bruk av flere parallelle assayer, løse dette problemet, men grunnet den korte prosjektperiode var det ikke mulig å undersøke dette. Den gjennomførte metyleringsanalysen fra prosjektet kan brukes som grunnlag for å etablere flere assayer.
Prosjektresultatene viser også at det var mulig å lage en MS-qPCR assay, som måler metyleringsraten ved en spesifikk posisjon i lakselus. MS qPCR-assayer er en sensitiv metode for å kvantifisere CpG-metylering ved en spesifikk posisjon, som kan brukes med standardutstyr i mange laboratorier. Selv om posisjonen valgt viste seg ikke å være optimal siden den ikke kunne skille entydig lus fra de undersøkte gruppene (som beskrevet ovenfor) viste målinger utført med assayen signifikante forskjeller i metyleringsraten mellom voksne hunnlus samplet fra sjøørret sammenlignet med lus samplet fra oppdrettslaks ved den spesifikke posisjonen. Dette ble validert med ny innsamlete lus som hadde samme mønsteret i. Om det hadde vært en bias i de første prøvene som førte til metyleringsforskjeller i dette posisjonen mellom gruppene ville man ikke finne samme mønsteret i nye lus. Det kunne dessuten vises, at metyleringsraten ved den spesifikke posisjonen også kunne finnes igjen i avkommene til de målte voksne hunnlus. Forskjellene i kopepodittene var signifikant, men enda svakere. Sensitiviteten til assayen var ikke god nok til å måle metyleringsraten i enkelte kopepoditter, men bare i batcher av omtrent 100 kopepoditter (samplet fra samme eggstreng). Optimaliseringen av fikseringsmetoden til kopepodittene og til assayen kunne muligens løse dette problemet. Kopepodittene var i samsvar med de andre prøvene fiksert på RNA later. Bruk av RNA later var nødvendig for de voksne hunnlusene samplet i 2023 til bruk for samordnet DNA og RNA sekvensering, men DNA utbytte i etanolfikserte prøver er vanligvis bedre. Tidsrammen av prosjektet tillot ikke optimaliseringen av selve assayen. Selv om resultatene mellom lus samplet i 2023 og 2024 samsvarte, ville en videre validering av assayen med flere prøver (både voksne hunnlus og deres avkom) være nødvendig.
For å kunne anvende metoden trenges det videre utvikling som beskrevet ovenfor. Om dette ville føre i mål og metoden kunne anvendes på frittlevende kopepoditter i havet, kunne resultatene hjelpe til å verifisere resultater fra hydrodynamiske-biologiske modellene som blir brukt til å modellere spredning av lakseluslarver i havet. Kunnskap om spredning av luselarver kan blant annet hjelpe ved koordinering av avlusning eller ved plassering av merdene.
6.1 - Hovedfunn
• Der er signifikante forskjeller i DNA-metyleringsmønsteret og genuttrykket mellom lus samlet fra oppdrettslaks, sjøørret og røye.
• Selv om forskjellene ved de spesifikke posisjonene er signifikante på gruppenivået, er ingen av forskjellene entydige når man ser på individuelle lus. Bruk av flere markører kunne muligens løse dette problemet.
• En MS-qPCR assay, en assay som kan skille metyleringsraten ved en spesifikk posisjon, ble utviklet for en posisjon som ble funnet å ha forskjellig metyleringsrate i lus fra oppdrettslaks sammenlignet med lus fra sjøørret eller røye.
• Metyleringsraten målt med assayen viste signifikante forskjeller både i voksne hunnlus og i deres avkom, men metoden var under de brukte forholdene ikke sensitiv nok til å måle metyleringsraten i enkelte kopepoditter.
7 - Referanser
Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A and Mason CE (2012) methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology 13(10), R87. https://doi.org/10.1186/gb-2012-13-10-r87.
Asplin L, Albretsen J, Johnsen IA and Sandvik AD (2020) The hydrodynamic foundation for salmon lice dispersion modeling along the Norwegian coast. Ocean Dynamics 70(8), 1151-1167. https://doi.org/10.1007/s10236-020-01378-0.
Asplin L, Boxaspen K and Sandvik A (2004) Modelled distribution of sea lice in a Norwegian Fjord. ICES CM 2004/ P11. http://hdl.handle.net/11250/100605.
Beamish RJ, Neville CM, Sweeting RM and Ambers N (2005) Sea lice on adult Pacific salmon in the coastal waters of Central British Columbia, Canada. Fisheries Research 76(2), 198-208. https://doi.org/10.1016/j.fishres.2005.06.007.
Bøhn T, Nilsen R, Gjelland KØ, Biuw M, Sandvik AD, Primicerio R, Karlsen Ø and Serra-Llinares RM (2022) Salmon louse infestation levels on sea trout can be predicted from a hydrodynamic lice dispersal model. Journal of Applied Ecology 59(3), 704-714. https://doi.org/10.1111/1365-2664.14085.
Diaz Pauli B, Berntsen HH, Thorstad EB, Homrum Eì, Lusseau SM, Wennevik V and Utne KR (2022) Geographic distribution, abundance, diet, and body size of invasive pink salmon (Oncorhynchus gorbuscha) in the Norwegian and Barents Seas, and in Norwegian rivers. Ices Journal of Marine Science 80(1), 76-90. https://doi.org/10.1093/icesjms/ fsac224.
Eichner C, Frost P, Dysvik B, Jonassen I, Kristiansen B and Nilsen F (2008) Salmon louse (Lepeophtheirus salmonis) transcriptomes during post molting maturation and egg production, revealed using EST-sequencing and microarray analysis. BMC Genomics 9, 126. https://doi.org/10.1186/1471-2164-9-126.
Feil R and Fraga MF (2012) Epigenetics and the environment: emerging patterns and implications. Nature Reviews Genetics 13(2), 97-109. https://doi.org/10.1038/nrg3142.
Feng SH, Cokus SJ, Zhang XY, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M and Jacobsen SE (2010) Conservation and divergence of methylation patterning in plants and animals. Proceedings of the National Academy of Sciences of the United States of America 107(19), 8689-8694. https://doi.org/10.1073/pnas.1002720107.
Flaten AC, Davidsen JG, Thorstad EB, Whoriskey F, Rønning L, Sjursen AD, Rikardsen AH and Arnekleiv JV (2016) The first months at sea: marine migration and habitat use of sea trout Salmo trutta post-smolts. Journal of Fish Biology 89(3), 1624-1640. https://doi.org/10.1111/jfb.13065.
Friedman JH, Hastie T and Tibshirani R (2010) Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of Statistical Software 33(1), 1 - 22. https://doi.org/10.18637/jss.v033.i01.
Gjelland K, Serra-Llinares RM, Hedger RD, Arechavala-Lopez P, Nilsen R, Finstad B, Uglem I, Skilbrei OT and Bjørn PA (2014) Effects of salmon lice infection on the behaviour of sea trout in the marine phase. Aquaculture Environment Interactions 5(3), 221-233. https://doi.org/10.3354/aei00105.
Hamre LA, Bui S, Oppedal F, Skern-Mauritzen R and Dalvin S (2019) Development of the salmon louse Lepeophtheirus salmonis parasitic stages in temperatures ranging from 3 to 24°C. Aquaculture Environment Interactions 11, 429-443. https://doi.org/10.3354/aei00320.
Heberle H, Meirelles GV, da Silva FR, Telles GP and Minghim R (2015) InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. Bmc Bioinformatics 16(1), 169. https://doi.org/10.1186/s12859-015-0611-3.
Jensen AJ, Finstad B, Fiske P, Diserud OH and Thorstad EB (2020) Repeatable individual variation in migration timing in two anadromous salmonids and ecological consequences. Ecology and Evolution 10(20), 11727-11738. https://doi.org/10.1002/ece3.6808.
Jensen JLA, Rikardsen AH, Thorstad EB, Suhr AH, Davidsen JG and Primicerio R (2014) Water temperatures influence the marine area use of Salvelinus alpinus and Salmo trutta. Journal of Fish Biology 84(6), 1640-1653. https://doi.org/10.1111/jfb.12366.
Johnsen IA, Asplin LC, Sandvik AD and Serra-Llinares RM (2016) Salmon lice dispersion in a northern Norwegian fjord system and the impact of vertical movements. Aquaculture Environment Interactions 8, 99-116. https://doi.org/10.3354/aei00162.
Jones S (2011) Mechanisms of resistance among salmon to the parasitic copepod Lepeophtheirus salmonis. Journal of Aquaculture Research and Development 01. https://doi.org/10.4172/2155-9546.s2-003.
Keller TE, Han P and Yi SV (2015) Evolutionary Transition of Promoter and Gene Body DNA Methylation across Invertebrate–Vertebrate Boundary. Molecular Biology and Evolution 33(4), 1019-1028. https://doi.org/10.1093/molbev/msv345.
Klemetsen A, Amundsen P-A, Dempson JB, Jonsson B, Jonsson N, O'Connell MF and Mortensen E (2003) Atlantic salmon Salmo salar L., brown trout Salmo trutta L. and Arctic charr Salvelinus alpinus (L.): a review of aspects of their life histories. Ecology of Freshwater Fish 12(1), 1-59. https://doi.org/10.1034/j.1600-0633.2003.00010.x.
Klughammer J, Romanovskaia D, Nemc A, Posautz A, Seid CA, Schuster LC, Keinath MC, Lugo Ramos JS, Kosack L, Evankow A, Printz D, Kirchberger S, Ergüner B, Datlinger P, Fortelny N, Schmidl C, Farlik M, Skjærven K, Bergthaler A, Liedvogel M, Thaller D, Burger PA, Hermann M, Distel M, Distel DL, Kübber-Heiss A and Bock C (2023) Comparative analysis of genome-scale, base-resolution DNA methylation profiles across 580 animal species. Nat Commun 14(1), 232. https://doi.org/10.1038/s41467-022-34828-y.
Lee YH, Kim M-S, Wang M, Bhandari RK, Park HG, Wu RS-S and Lee J-S (2022) Epigenetic plasticity enables copepods to cope with ocean acidification. Nature Climate Change 12(10), 918-927. https://doi.org/10.1038/s41558-022-01477-4.
Lorentzen EA (2023) Fant lus også på pukkellaks. Available at https://www.hi.no/hi/nyheter/ 2023/november/fant-lus-ogsa-pa-pukkellaks (accessed 23.12.2024).
Morton AB and Williams R (2003) First report of a sea louse, Lepeophtheirus salmonis, infestation on juvenile Pink Salmon, Oncorhynchus gorbuscha, in nearshore habitat. Canadian Field-Naturalist 117(4), 634-641. https://doi.org/10.22621/cfn.v117i4.834.
Nilsen R, Serra-Llinares RM, Sandvik AD, Harvey A, Tonstad A, Uglem I, Lehmann GB and Karlsen Ø (2022) Lakselusinfestasjon på vill laksefisk langs Norskekysten i 2022. Available at https://www.hi.no/templates/reporteditor/report-pdf?id=66076&25333918
Nilsen R, Serra-Llinares RM, Sandvik AD, Harvey A, Tonstad A, Uglem I, Lehmann GB and Karlsen Ø (2023) Lakselusinfestasjon på vill laksefisk våren 2023. Available at https://www.hi.no/templates/reporteditor/report-pdf?id=72697&41004072.
Nilsen R, Serra Llinares RM, Ambjørndalen V, Berg M, Lehmann GB, Tonstad A, Finstad B and Karlsen Ø (2020) Havforskningsinstituttet Ruse og Garn NALO 2020. Available at https://doi.org/10.21335/NMDC-477241498.
Piferrer F and Wang H-P (2023) Epigenetics in aquaculture, 1st edn. Hoboken, NJ: John Wiley & Sons Inc.
Rune N, Rosa Maria S-L, Anne DS, Agnes MM, Alison H, Ingebrigt U, Gunnar BL and Ørjan K (2022) Lakselusinfestasjon på vill laksefisk langs Norskekysten i 2021. Available at https://www.hi.no/templates/reporteditor/report-pdf?id=53927&45161735.
Saito T, Espe M, Vikeså V, Bock C, Thomsen TH, Adam AC, Fernandes JMO and Skjaerven KH (2024) One-carbon metabolism nutrients impact the interplay between DNA methylation and gene expression in liver, enhancing protein synthesis in Atlantic salmon. Epigenetics 19(1), 2318517. https://doi.org/10.1080/15592294.2024.2318517.
Samsing F, Oppedal F, Dalvin S, Johnsen I, Vågseth T and Dempster T (2016) Salmon lice (Lepeophtheirus salmonis) development times, body size, and reproductive outputs follow universal models of temperature dependence. Canadian Journal of Fisheries and Aquatic Sciences 73(12), 1841-1851. https://doi.org/10.1139/cjfas-2016-0050.
Serra-Llinares RM, Bjorn PA, Finstad B, Nilsen R, Harbitz A, Berg M and Asplin L (2014) Salmon lice infection on wild salmonids in marine protected areas: an evaluation of the Norwegian 'National Salmon Fjords'. Aquaculture Environment Interactions 5(1), 1-16. https://doi.org/10.3354/aei00090.
Skern-Mauritzen R, Malde K, Eichner C, Dondrup M, Furmanek T, Besnier F, Komisarczuk AZ, Nuhn M, Dalvin S, Edvardsen RB, Klages S, Huettel B, Stueber K, Grotmol S, Karlsbakk E, Kersey P, Leong JS, Glover KA, Reinhardt R, Lien S, Jonassen I, Koop BF and Nilsen F (2021) The salmon louse genome: Copepod features and parasitic adaptations. Genomics 113(6), 3666-3680. https://doi.org/10.1016/j.ygeno.2021.08.002
Skjærven KH, Adam A-C, Saito T, Waagbø R and Espe M (2023) Epigenetics in Fish Nutritional Programming. In Epigenetics in Aquaculture. 231-244.
Thompson C, Bui S, Dalvin S and Skern-Mauritzen R (2023) Disentangling the key drivers of salmon louse Lepeophtheirus salmonis fecundity using multiyear field samples. Aquaculture Environment Interactions 15, 161-178. https://www.int-res.com/abstracts/ aei/v15/p161-178/.
Vandegehuchte MB, De Coninck D, Vandenbrouck T, De Coen WM and Janssen CR (2010) Gene transcription profiles, global DNA methylation and potential transgenerational epigenetic effects related to Zn exposure history in Daphnia magna. Environmental Pollution 158(10), 3323-3329. https://doi.org/10.1016/j.envpol.2010.07.023.
Vandegehuchte MB, Lemière F, Vanhaecke L, Vanden Berghe W and Janssen CR (2010) Direct and transgenerational impact on Daphnia magna of chemicals with a known effect on DNA methylation. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology 151(3), 278-285. https://doi.org/10.1016/j.cbpc.2009.11.007.
Vollset KW, Qviller L, Skår B, Barlaup BT and Dohoo I (2018) Parasitic sea louse infestations on wild sea trout: separating the roles of fish farms and temperature. Parasites & Vectors 11(1), 609. https://doi.org/10.1186/s13071-018-3189-6.
8 - Leveranser
Oppstartsmøte: gjennomført den 21. august 2023
To statusrapporter: levert den 15. desember 2023, levert den 14. juni 2024
Prosjektmøte: gjennomført den 1. februar 2024
Presentasjon ved nasjonal konferanse: presentert ved Havbruk 2024 den 22.- 24. oktober
Presentasjon ved internasjonal konferanse: presentert ved IOmics in Aquaculture, Embrapa, Palmas, Brazil den 3.-5. desember 2024
Populærvitenskapelig presentasjon: HI rapport